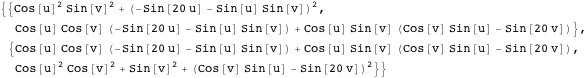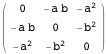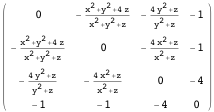This article shows how to use some of Mathematica’s built-in financial functions and define new functions useful for the practical analysis of real-world financial data. The main topics covered are linear programming and its application in bond portfolio management, conditional value-at-risk minimization, introductory time-series analysis, simulation, bootstrapping, robust equity portfolio optimization and artificial intelligence.
1. Introduction
Our main objective is to apply the Wolfram Language to solve financial models. We do not explain financial concepts or any mathematical background related to the financial applications introduced in this article. Nor do we introduce Mathematica. Wellin [1] gives a good introduction to programming in Mathematica. Mathematical background related to the models discussed here can be found in standard textbooks, including the ones cited.
First, we define three supporting functions used in the rest of this article.
The ![]() function downloads historical stock returns given its four arguments:
function downloads historical stock returns given its four arguments:
• a list of one or more ticker symbols
• the start date as a date object
• the end date as a date object
• the period as the frequency of data

The ![]() function computes 11 basic descriptive statistics given a list or a matrix of returns as its input argument.
function computes 11 basic descriptive statistics given a list or a matrix of returns as its input argument.

The ![]() function was taken from Stack Exchange Network (https://mathematica.stackexchange.com/questions/194234/plot-of-histogram-similar-to-output-from-risk) and modified. The function takes a vector of numerical data and returns a histogram with a handle on each side. You can drag these two thin vertical gray lines to vary the percentage of data within two values.
function was taken from Stack Exchange Network (https://mathematica.stackexchange.com/questions/194234/plot-of-histogram-similar-to-output-from-risk) and modified. The function takes a vector of numerical data and returns a histogram with a handle on each side. You can drag these two thin vertical gray lines to vary the percentage of data within two values.

Example
For example, this downloads monthly historical stock returns for Boeing Company (BA), Apple Inc. (AAPL) and NVIDIA Corporation (NVDA) for the period May 1, 2000, to May 30, 2019, and computes descriptive statistics. We chose returns of the Boeing Company for the histogram. Drag the handles at the far ends of the histogram (the thin vertical lines) to see the percentage of values that lie within two values.
![]()


The article is organized as follows:
• Section 2 introduces linear programming and applies it to bond portfolio management.
• Section 3 discusses mean-conditional value-at-risk portfolio optimization.
• Section 4 shows how to use built-in functions for introductory financial time-series analysis.
• Section 5 describes how simulation can be used in capital budgeting.
• Section 6 applies bootstrapping to risk management and financial planning.
• Section 7 describes robust equity portfolio optimization.
• Section 8 introduces functions useful for machine learning.
• Finally, the last section concludes the discussion.
2. Linear Programming and Applications
In this section, we illustrate a few applications of linear programming to financial problems similar to those in Cornuéjols, Peña and Tütüncü [2]. A linear program is an optimization problem whose purpose is to minimize or maximize a linear objective function subject to linear constraints. We first decide the decision variables, objective function and constraints. Then we find the values of the decision variables to optimize the objective function.
For vectors ![]() ,
, ![]() and
and ![]() and matrices
and matrices ![]() and
and ![]() , we can specify a general linear program in the following form:
, we can specify a general linear program in the following form:
 cx (objective function); subject to A x=b (equality constraints); D x>=d (inequality constraints); l<=x<=u (lower and upper bounds)")
We can convert any ![]() constraint to
constraint to ![]() by adding auxiliary variables.
by adding auxiliary variables.
The Wolfram Language has built-in functions to solve linear optimization problems with real variables. They include:
• ![]()
• ![]()
• ![]()
• ![]()
• ![]()
• ![]()
• ![]()
For large-scale problems, the most flexible and efficient of these is ![]() . The others are appropriate for solving linear programs written in terms of equations.
. The others are appropriate for solving linear programs written in terms of equations.
Example Using 
We consider an example very similar to the one presented in [2]. Assume that we have obligations to pay cash flows in the next eight years as shown in the following table. The first row shows years and the second row shows the amount of cash to be paid each year.
![]()
Also assume that we have five government bonds available to invest with the cash flows from the obligations and current prices given as follows.

To make a portfolio that minimizes the overall cost and still meets all the expected future cash payments, we can decide on how to allocate the funds by converting this problem into a linear program and solving it. Assume that ![]() is the number of bonds
is the number of bonds ![]() to purchase.
to purchase.
We define four variables for convenience.

![]()
![]()
![]()
![]()
![]()
Then the problem can be stated as follows.

This solves the problem.

![]()
Same Example Using 
The problem can also be solved using the ![]() function. One of its most useful forms is
function. One of its most useful forms is ![]() , where:
, where:
• ![]() is the vector of coefficients of the objective function.
is the vector of coefficients of the objective function.
• ![]() is the matrix of coefficients in the constraints.
is the matrix of coefficients in the constraints.
• ![]() is a two-column matrix representing the constants on the right side of the constraints and the direction of inequality.
is a two-column matrix representing the constants on the right side of the constraints and the direction of inequality.
![]()
• ![]() is a two-column matrix of lower and upper bounds for the decision variables.
is a two-column matrix of lower and upper bounds for the decision variables.
The variables ![]() and
and ![]() were defined before; here they are displayed in matrix form.
were defined before; here they are displayed in matrix form.
![]()

![]()

We define ![]() from
from ![]() and
and ![]() .
.
![]()

![]()
![]()
This solves the problem.

![]()
Mathematica’s built-in capabilities to solve linear programming problems can be used in a wide variety of financial problems. We refer interested readers to [2].
3. Mean-CVaR Optimization
In this section, we solve the mean-CVaR portfolio problem, which was proposed by Rockafellar and Uryasev [3]. CVaR optimization does not depend on any assumption of how returns are distributed, but it works for the normal distribution. We summarize the linear programming formulation of the CVaR problem.
Following [3], ![]() is the joint density function of the underlying asset returns
is the joint density function of the underlying asset returns ![]() , where
, where ![]() is the return on asset
is the return on asset ![]() ;
; ![]() is the loss associated with the decision vector
is the loss associated with the decision vector ![]() , where
, where ![]() is the proportion of money invested in asset
is the proportion of money invested in asset ![]() ; and
; and ![]() is the
is the ![]() -quantile of the loss distribution.
-quantile of the loss distribution.
Then the CVaR can be defined as:
![]() ,
,
where ![]() is expectation and
is expectation and ![]()
For a sample size ![]() , the CVaR is approximately:
, the CVaR is approximately:
![]() .
.
The problem can be restated as a linear optimization problem by introducing auxiliary variables, one for each observation in the sample:
Find ![]() ,
,
subject to ![]() and
and ![]() for
for ![]() .
.
As a linear program, the problem is:
Find ![]() ,
,
subject to
![]() and
and ![]() for
for ![]() ,
,
![]() and
and ![]() for
for ![]() ,
,
![]() ,
,
where ![]() ,
, ![]() and
and ![]() is the target optimal portfolio return.
is the target optimal portfolio return.
We define the function ![]() to estimate the optimal weights that minimize CVaR. It takes three arguments:
to estimate the optimal weights that minimize CVaR. It takes three arguments:
• the returns matrix (![]() )
)
• the target portfolio return (![]() )
)
• the confidence level (![]() ), between 0.9 and 0.99
), between 0.9 and 0.99

Example
This downloads monthly returns of three stocks over the period May 1, 2000, to May 30, 2019, and computes the CVaR-based optimal weights and associated values given the target portfolio return and confidence level.
![]()
![]()

The function computes optimal weights for a long-only portfolio. It can easily be modified to account for short-selling.
4. Introductory Time-Series Analysis and Forecasting
Data collected over time is common in finance. Mathematica has many built-in functions to model the stochastic nature of financial time series and to forecast the future value of a series. This section gives examples of functions that are useful for model specification, estimation, diagnostics and forecasting of univariate time-series data.
Constructing and Visualizing Time Series
The first step in any exploratory analysis is to construct and plot time series. You can use the built-in functions ![]() and
and ![]() to construct financial time series as pairs
to construct financial time series as pairs ![]() . There are two formats for each:
. There are two formats for each:
• ![]()
• ![]()
• ![]()
• ![]()
Time-series data can be manipulated using many built-in functions; see the documentation.
![]()

Once we create a time series, we can use functions like ![]() or
or ![]() to visualize it.
to visualize it.
We illustrate the historical global price of WTI Crude (POILWTIUSDM). First, we download the historical price of WTI crude oil since January, 1990, and then make a plot. Use the API key 207071a5f2e90e7816259d3c32c1ab81 if needed.


Model Fitting and Its Diagnostics
The built-in function ![]() supports both linear and nonlinear time-series models. It fits an automatically selected parametric model to a time series. We can customize the model fit specification by changing its options. The currently supported families of models are:
supports both linear and nonlinear time-series models. It fits an automatically selected parametric model to a time series. We can customize the model fit specification by changing its options. The currently supported families of models are:
• autoregressive (AR)
• moving-average (MA)
• autoregressive moving-average (ARMA)
• autoregressive integrated moving-average (ARIMA)
• seasonal autoregressive moving-average (SARMA)
• seasonal integrated autoregressive moving-average (SARIMA)
• autoregressive conditionally heteroscedastic (ARCH)
• generalized autoregressive conditionally heteroscedastic (GARCH)
You can find descriptions of these models in any time-series books, including Tsay [4].
Although ![]() selects the model automatically, there are many built-in functions for choosing appropriate values for the parameterizations for a given family and checking the appropriateness of the fitted models.
selects the model automatically, there are many built-in functions for choosing appropriate values for the parameterizations for a given family and checking the appropriateness of the fitted models.
Next, we are going to show the use of some tools for model specification and checking the adequacy of fitted models.
Use ![]() to test whether the data is autocorrelated. (Use
to test whether the data is autocorrelated. (Use ![]() to estimate the partial correlation function of the data.)
to estimate the partial correlation function of the data.)
![]()
![]()
Use ![]() to test whether data comes from an autoregressive time-series process with unit root.
to test whether data comes from an autoregressive time-series process with unit root.
![]()

A number of other tools are available for model specification:
• Akaike information criterion (AIC)
• Finite sample corrected AIC (AICc)
• Bayesian information criterion (BIC)
• Schwartz–Bayes information criterion (SBC)
To choose the appropriate model for the ![]() , we can do the following. (For information on
, we can do the following. (For information on ![]() , see the Mathematica help system.)
, see the Mathematica help system.)
![]()

Once the model has been specified, you can estimate its parameters with ![]() and you can assess its goodness of fit through analysis of residuals.
and you can assess its goodness of fit through analysis of residuals.
This estimates the parameters of the model for ![]() .
.
![]()
![]()
![]()
Use ![]() ,
, ![]() or
or ![]() of the residuals to assess the whiteness of the model residuals.
of the residuals to assess the whiteness of the model residuals.
![]()

Forecasting
A primary objective of building a time-series model is to forecast its future values. Prediction limits are important to assess the potential accuracy of the forecast. We can use ![]() to forecast unobserved future values. The function takes the methods
to forecast unobserved future values. The function takes the methods ![]() ,
, ![]() and
and ![]() . We use mean-squared errors to get the precision of our prediction.
. We use mean-squared errors to get the precision of our prediction.
This calculates and plots the forecast for the next 5 months of the series within 95% confidence limits.


ARCH and GARCH Models
The autoregressive conditional heteroskedasticity (ARCH) model and the generalized ARCH model (GARCH) are often used to get a volatility forecast of a time series. You can also use the built-in function ![]() to estimate parameters of these volatility models. Most volatility models are based on using returns that are obtained after subtracting unconditional mean returns. For our illustration, we de-mean our returns data. The parameters of the model are typically estimated with maximum likelihood.
to estimate parameters of these volatility models. Most volatility models are based on using returns that are obtained after subtracting unconditional mean returns. For our illustration, we de-mean our returns data. The parameters of the model are typically estimated with maximum likelihood.
To estimate a GARCH model for WTI Crude data, we find the de-mean data and estimate the model as follows.
![]()

You can use the built-in functions ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() and
and ![]() to simulate time-series data and for risk management.
to simulate time-series data and for risk management. ![]() is another important function to estimate the parameters of a process given its data and model specification.
is another important function to estimate the parameters of a process given its data and model specification.
5. Simulating Data and Financial Application
A financial model consisting of fixed relations and variables may not be accurate because most relationships between financial variables are random. Therefore, we must be able to incorporate stochasticity. Monte Carlo simulation is widely used to represent the true features of random modeling. Simulation modeling is a computer-based modeling technique that mimics a real-life situation and helps to incorporate uncertainties in input variables. Such techniques give a distribution of a forecast variable, not just a single value. Therefore, it is very useful when we are uncertain about future outcomes. In this section, we give examples for simulating data using Mathematica and show an application in capital budgeting.
![]() is a powerful function to get data from built-in statistical distributions, including those that are:
is a powerful function to get data from built-in statistical distributions, including those that are:
• continuous or discrete
• univariate or multivariate
• parametric or derived
• defined by data
Example in Project Risk Analysis
Consider a situation in which you have to evaluate an investment by forecasting the present value of its future cash flows.
We define a function ![]() to compute the present value of future cash flows. Here are its 11 arguments and values for an example. Assume that the revenue and terminal value both follow a triangular distribution and that gross margin follows a uniform distribution.
to compute the present value of future cash flows. Here are its 11 arguments and values for an example. Assume that the revenue and terminal value both follow a triangular distribution and that gross margin follows a uniform distribution.


For the given values, we simulate the data 10,000 times. (This takes a minute or so.)
![]()
We can summarize the data as follows.
![]()

Here is a histogram of the distribution of the present value of the cash flows. Drag the handles (thin vertical lines) on either side of the red region to see the percentage of cash flows that falls within the range of data.
![]()

This is just one example of simulation. We can define similar functions to compute the value of a firm using different valuation models:
• discounted cash flows
• residual operating income valuation
• abnormal growth in earnings valuation
There are many other areas of finance where simulation can be used.
6. Bootstrapping and Financial Application
We can apply the bootstrap approach in several contexts in finance. When the data is limited and the true distribution of the population is unknown, we can generate the sampling distribution of a statistic by generating many new samples from the original data and use the empirical distribution for statistical inference. This is called bootstrapping. Performing a bootstrap analysis in Mathematica is very straightforward using the ![]() or
or ![]() functions with or without replacement.
functions with or without replacement.
Performing bootstrap analysis entails two steps. First, we define a function that computes the statistic of interest. Second, we estimate the statistic of interest by repeatedly sampling observations (usually 10,000 times or more) from the original sample with replacement. Then we can use the distribution of sample statistics to infer an appropriate decision.
In this section, we illustrate the use of bootstrapping through two examples.
Example 1
We consider estimating the distribution of an equally weighted portfolio’s conditional value-at-risk (CVaR) using weekly returns of Walmart stock (WMT) and Procter & Gamble (PG) over the period January 1, 1982, to March 30, 2019.
Step 1
We get the historical weekly returns data.
![]()
Step 2
We define two functions.
The function ![]() computes the conditional value-at-risk given the returns data.
computes the conditional value-at-risk given the returns data.
![]()
The function ![]() returns a distribution of conditional value-at-risk measures given the size of the sample.
returns a distribution of conditional value-at-risk measures given the size of the sample.

We use these functions to get distributions of CVaR with 10,000 observations.
![]()
We summarize the data.


Example 2
The next example focuses on retirement planning using the bootstrapping concept. Assume that we want to calculate the terminal value of the following retirement portfolio. The savings are invested equally in two market indices: the S&P 500 Index and the NASDAQ 100 Index. Assume that future returns would be a random draw from past returns. The initial deposit is $1000. Monthly saving for the next 20 years is $1,500. The number of retirement years is 15. During the 15 years of retirement, $2000 will be withdrawn monthly. Starting at the 10th year, $30,000 will be withdrawn annually for three years.
Define the function ![]() , which calculates the terminal value of a retirement portfolio. It takes 10 arguments:
, which calculates the terminal value of a retirement portfolio. It takes 10 arguments:
• returns of the stocks in which money is equally invested (![]() )
)
• initial portfolio value (![]() ), which must be positive
), which must be positive
• ![]() –years in service
–years in service
• ![]() –periodic saving
–periodic saving
• ![]() –frequency of contribution per year; coded as 12 for monthly data and 1 for annual data
–frequency of contribution per year; coded as 12 for monthly data and 1 for annual data
• ![]() –number of retirement years
–number of retirement years
• ![]() –periodic income during retirement years
–periodic income during retirement years
• ![]() –big annual withdraw amount during planning period
–big annual withdraw amount during planning period
• ![]() –big withdrawal starting year; annual and in successive years with no gap
–big withdrawal starting year; annual and in successive years with no gap
• ![]() –number of annual big withdrawals
–number of annual big withdrawals

Using the values given, this computes the terminal value.
![]()
![]()
![]()
7. Mean-Variance Optimization under Uncertainty
Optimization under uncertainty (or robust optimization) is another approach that helps to get solutions that are good for most realizations of data. Many financial problems fit into this framework; for example, the mean-variance portfolio optimization problem. Here uncertainty may arise due to many factors:
• uncertainty in the mean vector
• fluctuations in the covariation matrix
• variability of risk in the market over time
• imprecise model approximation
Feasibility depends on both the decision variable ![]() and the uncertain vector
and the uncertain vector ![]() ; uncertainty can be introduced in the expected value, the variance or both.
; uncertainty can be introduced in the expected value, the variance or both.
This section considers a mean-variance problem that allows some degree of variation in returns and covariances.
The standard form of a mean-variance problem is expressed in terms of the information about the expected returns and the covariance structure of the returns. For given asset returns ![]() , where
, where ![]() is the return on asset
is the return on asset ![]() ; and the decision vector
; and the decision vector ![]() , where
, where ![]() is the proportion of money invested in asset
is the proportion of money invested in asset ![]() ; the mean variance problem is written as:
; the mean variance problem is written as:
![]()
![]()
subject to ![]() .
.
Here  , where
, where ![]() is the covariance between securities
is the covariance between securities ![]() and
and ![]() ;
; ![]() is the risk coefficient;
is the risk coefficient;  , where
, where ![]() is the average return on security
is the average return on security ![]() ; and
; and ![]() is an
is an ![]() column vector of ones,
column vector of ones, ![]() .
.
It is important to find the portfolio with the maximum Sharpe ratio, which can be obtained by solving the following problem; assume that ![]() and denote the risk-free rate by
and denote the risk-free rate by ![]() :
:
![]()
![]()
subject to ![]() .
.
The analytical solution to this basic problem for portfolio optimal portfolio weights ![]() is
is
![]() .
.
We define the function ![]() to compute
to compute ![]() . It takes the arguments returns data (
. It takes the arguments returns data (![]() ) and risk-free rate (
) and risk-free rate (![]() ) and returns an optimal weight vector.
) and returns an optimal weight vector.

One way to get a robust solution to the mean-variance problem is to sample data in several scenarios to estimate parameters. Assuming that the returns have multivariate normal distribution, we define a function ![]() to compute tangency portfolios with simulated data. The function takes four arguments:
to compute tangency portfolios with simulated data. The function takes four arguments:
• ![]() –returns data
–returns data
• ![]() –risk-free rate
–risk-free rate
• ![]() –sample size
–sample size
• ![]() –number of iterations
–number of iterations
It returns a distribution of optimal Sharpe ratios.

We apply those two functions to simulated data and get the distribution of portfolio means that maximize the Sharpe ratio.
We download historical monthly returns as before, compute the distribution of maximum Sharpe ratios and draw a histogram.
![]()
![]()

Another way to get robustness in the parameter estimate is to introduce some kinds of uncertainty; interval uncertainty sets and ellipsoidal uncertainty sets are the most commonly used. Following Kim, Kim and Fabozzi [5] (using ![]() and
and ![]() instead of their
instead of their ![]() and
and ![]() ), the interval uncertainty set for the expected returns can be defined as:
), the interval uncertainty set for the expected returns can be defined as:
![]() ,
,
where ![]() , the variable
, the variable ![]() is an estimate of the expected return and
is an estimate of the expected return and ![]() is a constant used to control the expected returns of stock
is a constant used to control the expected returns of stock ![]() .
.
The mean-variance problem with box uncertainty can be written as:
![]()
![]()
![]()
subject to ![]() and where
and where ![]() is such that
is such that ![]() for
for ![]() .
.
This objective function can be modified as follows (see [5] for the derivation and explanation of the notation ![]() and
and ![]() ):
):
![]()
![]()
subject to ![]() and
and ![]() , where
, where ![]() is the transformation matrix
is the transformation matrix ![]() for
for ![]() a unit matrix of size
a unit matrix of size ![]() ; for example, when
; for example, when ![]() ,
, ![]() .
.
We define the function ![]() to find the solution to the mean variance optimization problem with box uncertainty. This function takes three arguments:
to find the solution to the mean variance optimization problem with box uncertainty. This function takes three arguments:
• ![]() –a matrix of stock returns
–a matrix of stock returns
• ![]() –the risk coefficient
–the risk coefficient
• ![]() –the confidence level for the uncertainty set
–the confidence level for the uncertainty set

We compute optimal weights for the following portfolio.
![]()
![]()

In [5], an ellipsoidal uncertainty set on expected returns is defined as:
![]() ,
,
where ![]() is the covariance matrix of estimation error of expected returns and
is the covariance matrix of estimation error of expected returns and ![]() controls the size of the ellipsoid. With this uncertainty set, the mean variance problem can be written as:
controls the size of the ellipsoid. With this uncertainty set, the mean variance problem can be written as:
![]()
![]() ,
,
subject to ![]() .
.
The covariance matrix of estimation errors can be approximated in several ways using the sample covariance matrix of stock returns. Assuming that the samples of stock returns are independent and identically distributed, [5] defines ![]() , where
, where ![]() is the covariance matrix of stock returns and
is the covariance matrix of stock returns and ![]() is the sample size.
is the sample size.
Define ![]() to compute an optimal portfolio with an ellipsoidal uncertainty. It takes three arguments:
to compute an optimal portfolio with an ellipsoidal uncertainty. It takes three arguments:
• returns (![]() )
)
• value of risk coefficient (![]() )
)
• confidence level for the uncertainty set (![]() )
)

We compute optimal weights for our usual portfolio subject to an ellipsoidal uncertainty set.
![]()
![]()

We can use the built-in function ![]() to optimize the portfolio problem, introducing uncertainty in the mean returns or in the covariance.
to optimize the portfolio problem, introducing uncertainty in the mean returns or in the covariance.


![]()
Similarly, we can introduce uncertainty in risk and solve the optimization problem using this same function. See the documentation for an example.
8. Artificial Intelligence
With increasing computational resources and larger datasets, machine learning or artificial intelligence is a growing field in finance. A recent book by Dixon, Halperin and Bilokon [6] is a good reference on theory and applications.
Although the Wolfram Language includes a wide range of functions that work on many types of data, including numerical, categorical, time series, textual, image and audio, we focus on the ![]() function and time-series data only. The
function and time-series data only. The ![]() function uses input data and returns a predictor function that can be used to forecast the value of dependant variables given the values of independent variables. In this section, we show two examples using financial time series.
function uses input data and returns a predictor function that can be used to forecast the value of dependant variables given the values of independent variables. In this section, we show two examples using financial time series.
One Predictor for Stock Returns
This example uses the Aruoba–Diebold–Scotti (ADS) business conditions index as one predictor of percentage change in the S&P 500 index. We define the function ![]() . It downloads the ADS index from the Federal Reserve Bank of Philadelphia and the S&P 500 index returns and then merges them for use in the
. It downloads the ADS index from the Federal Reserve Bank of Philadelphia and the S&P 500 index returns and then merges them for use in the ![]() function, given the start and end date as arguments.
function, given the start and end date as arguments.

Using this function, we download the data for the period January 30, 1970, to September 30, 2019, and generate a prediction function. (This takes several minutes.)
![]()
Once the prediction function ![]() is generated, we can use it to predict the future value of the stock index value. For instance, here it predicts percentage change in the S&P 500 index if ADS is 0.9, 2 or -1.2.
is generated, we can use it to predict the future value of the stock index value. For instance, here it predicts percentage change in the S&P 500 index if ADS is 0.9, 2 or -1.2.
![]()
![]()
Several Predictors for Stock Returns
In this example, we use five monthly macro variables to predict percentage change in the value of the S&P 500 index:
• USSLIND–the leading index for the United States
• UMCSENT–the University of Michigan consumer sentiment index
• CFNAIMA3–the Chicago Fed national activity index: three-month moving average
• MICH–the University of Michigan inflation expectation index
• T10Y2YM–10-year Treasury constant maturity minus 2-year Treasury constant maturity
The function ![]() takes a list of macroeconomic series IDs (
takes a list of macroeconomic series IDs (![]() ), start date and end date as input arguments. It returns values for specified macroeconomic variables and S&P 500 index returns in the format suitable for the
), start date and end date as input arguments. It returns values for specified macroeconomic variables and S&P 500 index returns in the format suitable for the ![]() function. The Federal Reserve Bank of St. Louis may require the API key to download its data. The API key can be obtained freely by creating a user account at https://fred.stlouisfed.org (click “my account” and follow the instructions). Use the API key 207071a5f2e90e7816259d3c32c1ab81 if needed.
function. The Federal Reserve Bank of St. Louis may require the API key to download its data. The API key can be obtained freely by creating a user account at https://fred.stlouisfed.org (click “my account” and follow the instructions). Use the API key 207071a5f2e90e7816259d3c32c1ab81 if needed.


Using this function, we download five macro variables as well as the S&P 500 index returns over the period January 30, 1983, to May 30, 2019.
![]()
We can generate the prediction function using the data and predict the value. ![]() can take the
can take the ![]() option to specify which regression method to use.
option to specify which regression method to use.
![]()
![]()
We have shown some applications of only the built-in function ![]() . However, the Wolfram Language comes with many other built-in functions that are useful in classification, discriminant analysis and neural networks. You can use these tools to learn from the data and build models to extract useful information. We encourage you to explore more about machine learning in Mathematica.
. However, the Wolfram Language comes with many other built-in functions that are useful in classification, discriminant analysis and neural networks. You can use these tools to learn from the data and build models to extract useful information. We encourage you to explore more about machine learning in Mathematica.
9. Conclusion
As financial data becomes increasingly available, serious data analysis requires knowing software to manipulate large datasets. Aside from demonstrating many built-in functions, we introduced many custom functions especially designed for technical computation of financial data. Mathematica can do much more than what we have shown in this article. The Wolfram Language in general and Mathematica in particular are well-suited to implement sophisticated financial models, including pricing securities, trading strategies, simulation, optimization, risk management and time-series analysis [7, 8]. Mathematica’s built-in knowledge is also very useful for asset pricing models based on estimating the stochastic discount factor using the generalized method of moments.
References
| [1] | P. Wellin, Essentials of Programming in Mathematica, Cambridge, UK: Cambridge University Press, 2016. |
| [2] | G. Cornuéjols, J. Peña and R. Tütüncü, Optimization Methods in Finance, 2nd ed., New York: Cambridge University Press, 2018. |
| [3] | R. T. Rockafellar and S. Uryasev, “Optimization of Conditional Value-at-Risk”, The Journal of Risk, 2(3), 2000 pp. 21–41. https://doi.org/10.21314/JOR.2000.038. |
| [4] | R. S. Tsay, An Introduction to Analysis of Financial Data with R, Hoboken, NJ: Wiley, 2013. |
| [5] | W. C. Kim, J. H. Kim and F. J. Fabozzi, Robust Equity Portfolio Management, Hoboken, NJ: Wiley, 2016. |
| [6] | M. Dixon, I. Halperin and P. Bilokon, Machine Learning in Finance from Theory to Practice, Cham, Switzerland: Springer, 2020. |
| [7] | A. L. Lewis, Option Valuation under Stochastic Volatility: With Mathematica Code, Newport Beach, CA: Finance Press, 2000. |
| [8] | A. L. Lewis, Option Valuation under Stochastic Volatility II: With Mathematica Code, Newport Beach, CA: Finance Press, 2016. |
| R. Adhikari, “Selected Financial Applications,” The Mathematica Journal, 2021. doi.org/10.3888/tmj.23-5. | |
About the Author
Ramesh Adhikari is an Associate Professor of Finance at Humboldt State University. Prior to coming to HSU, he taught undergraduate and graduate students at Tribhuvan University and worked at the Central Bank of Nepal. He was also a research fellow at Osaka Sangyo University, Osaka, Japan. He earned a Ph.D. in Financial Economics from the University of New Orleans. He is interested in the areas of computational finance and high-dimensional statistics.
Ramesh Adhikari
School of Business, Humboldt State University
1 Harpst Street
Arcata, CA 95521
ramesh.adhikari@humboldt.edu


















































































![(dem60[1]-(b1 ind60[1]+u1[1]))^2+
(dem60[2]-(b1 ind60[2]+u1[2]))^2+
...+
(dem60[n]-(b1 ind60[n]+u1[n]))^2](https://content.wolfram.com/sites/19/2020/12/Oldenburg_Image_2.gif "(dem60[1]-(b1 ind60[1]+u1[1]))^2+
(dem60[2]-(b1 ind60[2]+u1[2]))^2+
...+
(dem60[n]-(b1 ind60[n]+u1[n]))^2")




































































































![InterpretationBox[Description, TextCell[Enter column head here, Italic, Editable -> True]] InterpretationBox[Models for r-rf, TextCell[Enter column head here, Italic, Editable -> True]]; InterpretationBox[Capital asset pricing model (CAPM), TextCell[Enter data here, Editable -> True]] CAPM=alpha+beta_(MKT)MKT; InterpretationBox[Fama-French three-factor model, TextCell[Enter data here, Editable -> True]] InterpretationBox[{{FF, _, DocumentationBuild`Utils`Private`Parenth[3]}, =, {CAPM, +, {{beta, _, DocumentationBuild`Utils`Private`Parenth[SMB]}, SMB}, +, {{beta, _, DocumentationBuild`Utils`Private`Parenth[HML]}, HML}}}, TextCell[Enter data here, Editable -> True]]; InterpretationBox[Carhart four-factor model, TextCell[Enter data here, Editable -> True]] InterpretationBox[{{C, _, DocumentationBuild`Utils`Private`Parenth[4]}, =, {{FF, _, DocumentationBuild`Utils`Private`Parenth[3]}, +, {{beta, _, DocumentationBuild`Utils`Private`Parenth[RMW]}, RMW}}}, TextCell[Enter data here, Editable -> True]]; InterpretationBox[Fama-French five-factor model, TextCell[Enter data here, Editable -> True]] InterpretationBox[{{FF, _, DocumentationBuild`Utils`Private`Parenth[5]}, =, {{C, _, DocumentationBuild`Utils`Private`Parenth[4]}, +, {{beta, _, DocumentationBuild`Utils`Private`Parenth[CMA]}, CMA}}}, TextCell[Enter data here, Editable -> True]]](https://content.wolfram.com/sites/19/2020/08/Adhikari-1_Image_1.gif "InterpretationBox[Description, TextCell[Enter column head here, Italic, Editable -> True]] InterpretationBox[Models for r-rf, TextCell[Enter column head here, Italic, Editable -> True]]; InterpretationBox[Capital asset pricing model (CAPM), TextCell[Enter data here, Editable -> True]] CAPM=alpha+beta_(MKT)MKT; InterpretationBox[Fama-French three-factor model, TextCell[Enter data here, Editable -> True]] InterpretationBox[{{FF, _, DocumentationBuild`Utils`Private`Parenth[3]}, =, {CAPM, +, {{beta, _, DocumentationBuild`Utils`Private`Parenth[SMB]}, SMB}, +, {{beta, _, DocumentationBuild`Utils`Private`Parenth[HML]}, HML}}}, TextCell[Enter data here, Editable -> True]]; InterpretationBox[Carhart four-factor model, TextCell[Enter data here, Editable -> True]] InterpretationBox[{{C, _, DocumentationBuild`Utils`Private`Parenth[4]}, =, {{FF, _, DocumentationBuild`Utils`Private`Parenth[3]}, +, {{beta, _, DocumentationBuild`Utils`Private`Parenth[RMW]}, RMW}}}, TextCell[Enter data here, Editable -> True]]; InterpretationBox[Fama-French five-factor model, TextCell[Enter data here, Editable -> True]] InterpretationBox[{{FF, _, DocumentationBuild`Utils`Private`Parenth[5]}, =, {{C, _, DocumentationBuild`Utils`Private`Parenth[4]}, +, {{beta, _, DocumentationBuild`Utils`Private`Parenth[CMA]}, CMA}}}, TextCell[Enter data here, Editable -> True]]")




































![InterpretationBox[Greeks, TextCell[Enter data here, Editable -> True]] Evaluation InterpretationBox[Interpretation, TextCell[Enter data here, Editable -> True]]; InterpretationBox[estimated change in value relative to change in:, TextCell[Enter data here, Editable -> True]]; InterpretationBox[Delta (Delta), TextCell[Enter data here, Editable -> True]] (partialV)/(partialS) InterpretationBox[the underlying asset price, TextCell[Enter data here, Editable -> True]]; InterpretationBox[Vega (upsilon), TextCell[Enter data here, Editable -> True]] (partialV)/(partialsigma) InterpretationBox[the underlying asset volatility, TextCell[Enter data here, Editable -> True]]; InterpretationBox[Rho (rho), TextCell[Enter data here, Editable -> True]] (partialV)/(partialr) InterpretationBox[the risk-free interest rate, TextCell[Enter data here, Editable -> True]]; Theta (theta) (partialV)/(partialT) the time to expiration (for a negative change); Gamma (Gamma) (partial^2V)/(partialS^2) estimated change in delta relative to a change in the underlying asset price](https://content.wolfram.com/sites/19/2020/08/Adhikari-1_Image_38.gif "InterpretationBox[Greeks, TextCell[Enter data here, Editable -> True]] Evaluation InterpretationBox[Interpretation, TextCell[Enter data here, Editable -> True]]; InterpretationBox[estimated change in value relative to change in:, TextCell[Enter data here, Editable -> True]]; InterpretationBox[Delta (Delta), TextCell[Enter data here, Editable -> True]] (partialV)/(partialS) InterpretationBox[the underlying asset price, TextCell[Enter data here, Editable -> True]]; InterpretationBox[Vega (upsilon), TextCell[Enter data here, Editable -> True]] (partialV)/(partialsigma) InterpretationBox[the underlying asset volatility, TextCell[Enter data here, Editable -> True]]; InterpretationBox[Rho (rho), TextCell[Enter data here, Editable -> True]] (partialV)/(partialr) InterpretationBox[the risk-free interest rate, TextCell[Enter data here, Editable -> True]]; Theta (theta) (partialV)/(partialT) the time to expiration (for a negative change); Gamma (Gamma) (partial^2V)/(partialS^2) estimated change in delta relative to a change in the underlying asset price")