We describe the use of classical and metric multidimensional scaling methods for graphical representation of the proximity between collections of data consisting of cases characterized by multidimensional attributes. These methods can preserve metric differences between cases, while allowing for dimensional reduction and projection to two or three dimensions ideal for data exploration. We demonstrate these methods with three datasets for: (i) the immunological similarity of influenza proteins measured by a multidimensional assay; (ii) influenza protein sequence similarity; and (iii) reconstruction of airport-relative locations from paired proximity measurements. These examples highlight the use of proximity matrices, eigenvalues, eigenvectors, and linear and nonlinear mappings using numerical minimization methods. Some considerations and caveats for each method are also discussed, and compact Mathematica programs are provided.
1. Introduction
One “curse” of high- and multidimensional data is the difficulty in graphically displaying how the data points are related to each other. Each data point (in some fields referred to as a case) is characterized by a vector of attributes (e.g. shape, color, temperature, etc.) with numerical values that form a set of coordinates. These coordinates specify a location in an ![]() -dimensional space, with
-dimensional space, with ![]() being the number of variables used to describe each case. Visualization of such
being the number of variables used to describe each case. Visualization of such ![]() -dimensional data is problematic as, for example, we do not think in 17 dimensions! For human comprehension, we are limited to two- or three-dimensional graphics, sometimes referred to as visualizable dimensions, in which each case is represented by a data point. The distances between points and their relative locations reflect their proximity (similarity or dissimilarity), as measured by a metric function of their attributes, such as Euclidean distance. Larger distances between cases indicate that they are less related. Fortunately, multidimensional scaling (MDS) methods can be used to visualize the degree of proximity for high-dimensional datasets [1, 2, 3]. MDS methods can be used to project the
-dimensional data is problematic as, for example, we do not think in 17 dimensions! For human comprehension, we are limited to two- or three-dimensional graphics, sometimes referred to as visualizable dimensions, in which each case is represented by a data point. The distances between points and their relative locations reflect their proximity (similarity or dissimilarity), as measured by a metric function of their attributes, such as Euclidean distance. Larger distances between cases indicate that they are less related. Fortunately, multidimensional scaling (MDS) methods can be used to visualize the degree of proximity for high-dimensional datasets [1, 2, 3]. MDS methods can be used to project the ![]() -dimensional coordinates of each case to two or three dimensions, while preserving the relative distances between cases [4, 5]. These methods are increasingly employed in such diverse fields as immunology, molecular biology, marketing, perceptual mapping, and ecology to visualize relative “distances” between viruses, molecules, species, and customers’ behavior. In this article, we discuss some mathematical details of classical and metric MDS methods and build generalizable Mathematica programs that can be used for each. We incorporate use cases from geography, molecular virology, and immunology.
-dimensional coordinates of each case to two or three dimensions, while preserving the relative distances between cases [4, 5]. These methods are increasingly employed in such diverse fields as immunology, molecular biology, marketing, perceptual mapping, and ecology to visualize relative “distances” between viruses, molecules, species, and customers’ behavior. In this article, we discuss some mathematical details of classical and metric MDS methods and build generalizable Mathematica programs that can be used for each. We incorporate use cases from geography, molecular virology, and immunology.
Before covering the analytical details, it is worth briefly discussing the variety of MDS methods. The simplest method is referred to as classical multidimensional scaling, also known as principal coordinate analysis (PCoA) [1]. PCoA is not to be confused with principal component analysis (PCA). The difference between PCA and classical MDS/PCoA is primarily based on the input data. PCA starts with a set of cases or data points ![]() , each described by a set of attributes
, each described by a set of attributes ![]() , and uses a Euclidean distance to project the data to a lower-dimensional space oriented to maximize the variance observed between data points. In contrast, classical MDS begins with an input matrix that specifies proximities between pairs of items, often referred to as dissimilarities. It outputs a coordinate matrix in a lower dimension than the starting data (e.g. 120 dimensions projected to two) that preserves the relative distances between all data points. Classical MDS was originally developed by Gower [1] and others and is often demonstrated with a simple problem: given only the distances between entities (e.g. cities), is it possible to calculate the map showing their relative distances? We use this example in Section 4. In the specific case where the dissimilarity is measured by a Euclidean distance, PCA and classical MDS/PCoA are mathematically identical to another dimensional reduction method, singular value decomposition (SVD). In that case, there may be computational advantages to using SVD, which are discussed in this article.
, and uses a Euclidean distance to project the data to a lower-dimensional space oriented to maximize the variance observed between data points. In contrast, classical MDS begins with an input matrix that specifies proximities between pairs of items, often referred to as dissimilarities. It outputs a coordinate matrix in a lower dimension than the starting data (e.g. 120 dimensions projected to two) that preserves the relative distances between all data points. Classical MDS was originally developed by Gower [1] and others and is often demonstrated with a simple problem: given only the distances between entities (e.g. cities), is it possible to calculate the map showing their relative distances? We use this example in Section 4. In the specific case where the dissimilarity is measured by a Euclidean distance, PCA and classical MDS/PCoA are mathematically identical to another dimensional reduction method, singular value decomposition (SVD). In that case, there may be computational advantages to using SVD, which are discussed in this article.
While classical MDS relies on linear transformation of data using matrix operations, metric MDS methods depend on computational optimization. Metric MDS takes a proximity matrix and calculates a two- or three-dimensional coordinate matrix whose configuration minimizes the difference between the ![]() -dimensional and the calculated distances in the reduced dimensions for each case or data point [2, 3]. Thus, similar cases are located next to each other in a reduced-dimensional plot, while disparate cases are farther away, and the final coordinates can be projected to a planar or three-dimensional space. Other varieties of MDS, not discussed here, include nonmetric MDS, which uses isotonic regression methods, and generalized MDS, which projects the coordinates to an arbitrary smooth target space [3–7]. Metric MDS is more flexible than classical MDS for certain problems, including when the proximity matrix contains missing data or reflects a distance function that is not Euclidean. In addition, it can accommodate nonlinear mappings from the
-dimensional and the calculated distances in the reduced dimensions for each case or data point [2, 3]. Thus, similar cases are located next to each other in a reduced-dimensional plot, while disparate cases are farther away, and the final coordinates can be projected to a planar or three-dimensional space. Other varieties of MDS, not discussed here, include nonmetric MDS, which uses isotonic regression methods, and generalized MDS, which projects the coordinates to an arbitrary smooth target space [3–7]. Metric MDS is more flexible than classical MDS for certain problems, including when the proximity matrix contains missing data or reflects a distance function that is not Euclidean. In addition, it can accommodate nonlinear mappings from the ![]() -dimensional data space to the visualization. However, because metric MDS uses numerical optimization, it is computationally expensive. These tradeoffs are explored in Section 7.
-dimensional data space to the visualization. However, because metric MDS uses numerical optimization, it is computationally expensive. These tradeoffs are explored in Section 7.
A byproduct of both MDS methods is the proximity matrix, which can also be used for other high-dimensional visualizations, such as heat mapping and multidimensional clustering methods. Combining MDS with these methods often reveals different facets of the data and aids investigators in gaining understanding of the underlying data structure and exploratory data analysis.
In the following sections, we discuss construction of proximity matrices, classical MDS, and metric MDS. Several examples illustrate the strengths and pitfalls of these methods; the full datasets are included in the article. The tradeoff between computational efficiency and accuracy is touched on as well. Each section briefly covers the mathematical underpinning of the method, a step-by-step example, a self-contained function using compact code, considerations for the method and alternate approaches, and a synopsis of the most salient points. A separate section discusses accuracy and computational efficiency and compares these methods.
2. Computational Environment and Data
This article was created using Wolfram Mathematica Version 10.0.2, running Macintosh OS X 10.9.5 on a MacBook Pro with a 2.8 GHz Intel Core i7 processor with 16 GB RAM. It contains the entire code and all the data needed for execution of all examples. Internet connectivity is required to retrieve the airport distances and perform the mapping of the airport locations within Mathematica.
For the sake of aesthetics and clarity, we have collapsed cells containing code related to the display of tables and figures, which appear in their final form.
Execution of the last section of the article may take 4–8 minutes, given the number of minimization problems that are calculated. Also, earlier versions of Mathematica may not execute the GeoListPlot commands properly (or at all for Version 9.0 or lower), given that this functionality was added in Version 10.0 and several options were added between Versions 10.0 and 10.0.2.
3. Datasets and Display Functions
For the sake of brevity, the datasets are placed here in collapsed cells, along with several house-keeping functions for displaying tables and figures. Some of the data for intra-airport distances is retrieved by using the AirportData function and requires internet access if executing the
notebook again.
[collapsed cells]
4. Proximity Matrices
Classical and metric MDS methods rely on a symmetric input matrix ![]() (referred to as a proximity matrix) that specifies the
(referred to as a proximity matrix) that specifies the ![]() -dimensional distance between pairs of objects
-dimensional distance between pairs of objects ![]() and
and ![]() , denoted
, denoted ![]() . Proximity may be defined as a distance or dissimilarity.
. Proximity may be defined as a distance or dissimilarity.
In some applications, ![]() is composed of distances
is composed of distances ![]() that are specified directly, for example, the flying distances between airport pairs in the United States, as shown in Table 1. The proximity matrix is a symmetric distance matrix with the airport names in the first column and the corresponding airport abbreviations for the column labels.
that are specified directly, for example, the flying distances between airport pairs in the United States, as shown in Table 1. The proximity matrix is a symmetric distance matrix with the airport names in the first column and the corresponding airport abbreviations for the column labels.

4.1. Constructing a Proximity Matrix from a Cases by Attributes Table
Often, ![]() is not the primary data but must be calculated from a set of cases, each of which is described by a vector of attributes, with the primary data specified in an
is not the primary data but must be calculated from a set of cases, each of which is described by a vector of attributes, with the primary data specified in an ![]() cases by attributes data matrix. The attributes can be binary membership in a category or group, physical properties, or string sequences. The methods used to calculate
cases by attributes data matrix. The attributes can be binary membership in a category or group, physical properties, or string sequences. The methods used to calculate ![]() vary by the data and the application. The most commonly used metric is the Euclidean distance for continuous variables, but other distance functions may also be used, such as the Hamming and Manhattan distances, Boolean distances for Boolean data, Smith–Waterman string sequence similarity, or color distance functions for images.
vary by the data and the application. The most commonly used metric is the Euclidean distance for continuous variables, but other distance functions may also be used, such as the Hamming and Manhattan distances, Boolean distances for Boolean data, Smith–Waterman string sequence similarity, or color distance functions for images.
Table 2 is an example of such a cases by attributes matrix. The cases (rows) represent the influenza virus surface hemagglutinin proteins from various influenza strains (H1, H3, H5) and substrains (e.g. A/California/04/2009). The data is taken from an antibody binding assay, with the values reflecting the amount of antibody binding to each influenza hemagglutinin protein. The antibodies were isolated from the serum of ferrets infected with an influenza virus strain, shown in the columns. Each case (row) is characterized by a 15-dimensional attribute vector, with each attribute corresponding to the serum antibody binding values against that particular influenza strain hemagglutinin from a biological assay. Higher values indicate more antibody binding. Such assays are used to select vaccines each year against strains predicted to circulate in the fall. Note that there are 15 different cases characterized by 14 sera.
![]()

Before performing the MDS calculations, it is important to rescale each attribute to span the same range across all the cases, especially if the attributes have different units or variable ranges. This rescaling does not assume any specific statistical distribution. It is designed so that each attribute has equal weight in the proximity calculation, although weighted proximity matrices can also be used if the application requires this. Here we rescale the results for the above attributes so that the range of each attribute column (antibody binding test result for a single hemagglutinin) has a minimum of 0 and a maximum of 1000.
![]()

For multidimensional scaling, this data is transformed into a proximity matrix using a EuclideanDistance calculation. The larger the proximity measurement, the greater the difference between the hemagglutinin proteins from the two viral infection strains, as specified by the reactivity of immune sera against the probe hemagglutinin proteins. Table 4 uses the DistanceMatrix function to create a proximity matrix for the influenza. There are many ways to calculate distance matrices, and custom functions are often used. One convenient option is to use the DistanceMatrix function, which allows access to a number of standardized distance metrics (e.g. EuclideanDistance, ManhattanDistance, CorrelationDistance, etc.), which can be directly set by the option DistanceFunction. The DistanceMatrix function is contained in the HierarchicalClustering package.
![]()

4.2. Visualization
It is often helpful to visualize a proximity matrix as a heat map using the ArrayPlot function. In Figure 1 the most similar hemagglutinin protein pairs are blue and the most different are red. As can be seen, this representation of the data suggests a similarity of influenza virus hemagglutinin proteins from similar strains (H1, H3, H5) and the occurrence of year-to-year differences, especially for the
H1 strains.

Figure 1. Heat map of proximity matrix data.
4.3. Proximity Matrices from Molecular Sequence Comparisons
Another common application of multidimensional scaling is to compare protein or DNA sequences. Such sequences are essentially linear lists of strings that specify the protein or nucleic acid composition. Sequence differences are used to estimate molecular evolution, how the immune system may respond to a new virus after immunization, and cross-species comparison of molecules.
To construct a protein sequence proximity matrix, we begin with the amino acid sequences of the hemagglutinin proteins from influenza strains specified in the proximity matrix of the previous section. These were retrieved from the UniProt database (www.uniprot.org). Each letter in the sequence represents an amino acid, and each sequence is approximately 566 amino acids long.
To calculate the proximity matrix, we use a rescaled inverse of the Smith–Waterman similarity calculation, with the PAM70 similarity rules for amino acid comparison. Sequence similarity calculations have larger values with greater similarity, and the minimum score is generally the length of the sequence. Because MDS methods use proximity measures that increase monotonically with dissimilarity, the inverse of the sequence similarity function is used. Thus, for clarity in this example, we have transformed the result by subtracting a constant (528), which is one less than the minimum sequence length, and multiplying the inverse by a scale factor of ![]() . Note that nonzero values do
. Note that nonzero values do
not occur.


4.4. Synopsis: Proximity Matrices
The first step in multidimensional scaling is to create a proximity matrix. The value of each matrix element is a distance measure; the greater the difference between the two elements being compared, the greater the value of the metric. We have demonstrated three different cases of proximity matrix construction, including: (i) the airport distance case, where the proximities are known a priori and do not need to be calculated; (ii) the influenza protein antibody reactivity case, with proximity calculation from a cases by attributes matrix; and (iii) the influenza protein sequence example, with calculation from a sequence similarity measure. Heat maps are useful for displaying proximity matrices, giving a visual view of similarities. All the resulting proximity matrices can be used for MDS calculations, as will be discussed next.
5. Classical Multidimensional Scaling
Here we briefly describe the mathematics of classical multidimensional scaling, which uses a linear coordinate transformation calculation to reduce case coordinates from ![]() dimensions down to two or three dimensions. The approach begins with calculating a proximity matrix. The orientation of the new coordinate system is such that it accounts for the greatest amount of variance between cases, as defined by attributes, in the reduced number of dimensions. The new coordinates can be used to plot the relative locations of the data points and help visualize differences between data points and clusters of similar cases. The proximity matrix can also be used for data clustering and heat mapping, which provide alternate visualizations of case relationships.
dimensions down to two or three dimensions. The approach begins with calculating a proximity matrix. The orientation of the new coordinate system is such that it accounts for the greatest amount of variance between cases, as defined by attributes, in the reduced number of dimensions. The new coordinates can be used to plot the relative locations of the data points and help visualize differences between data points and clusters of similar cases. The proximity matrix can also be used for data clustering and heat mapping, which provide alternate visualizations of case relationships.
5.1. Mathematics
Let ![]() be a
be a ![]() matrix, where
matrix, where ![]() is the number of individual cases (e.g. data points) and
is the number of individual cases (e.g. data points) and ![]() is the number of measured variables or attributes (e.g. financial variables, consumer product attributes, antibody binding measurements, etc.), which are used to discriminate between each case
is the number of measured variables or attributes (e.g. financial variables, consumer product attributes, antibody binding measurements, etc.), which are used to discriminate between each case ![]() ,
, ![]() .
.
If the data attributes are of different scales and units, they are generally rescaled with a range of ![]() . This assures that each attribute contributes equally to the proximity measure.
. This assures that each attribute contributes equally to the proximity measure.
Alternatively, ![]() may remain in the original units if each attribute has the same range and units, or if a weighted proximity matrix is desired. Next, let the proximity matrix
may remain in the original units if each attribute has the same range and units, or if a weighted proximity matrix is desired. Next, let the proximity matrix ![]() of
of ![]() or
or ![]() rescaled be a matrix of dimensions
rescaled be a matrix of dimensions ![]() with elements
with elements
| (1) |
One can use a variety of applicable proximity measures (e.g. Euclidean, Mahalanobis, etc.) to create the proximity matrix. If the proximity measure is a Euclidean distance, then classical MDS is equivalent to principal component analysis (PCA). If any other distance function is used, then classical MDS will result in a different transformation that is not equivalent to PCA.
Let ![]() be the identity matrix of dimensions
be the identity matrix of dimensions ![]() and
and ![]() be the constant matrix of dimensions
be the constant matrix of dimensions ![]() , where
, where ![]() . Let
. Let ![]() be the matrix of the squared proximities:
be the matrix of the squared proximities:
| (2) |
The kernel matrix ![]() is derived by first calculating the matrix
is derived by first calculating the matrix ![]() from the identity matrix and the number of variables
from the identity matrix and the number of variables ![]() :
:
| (3) |
![]() is the matrix that double-centers the input matrix by subtracting the row and column means from every element of the matrix. Introducing the constant
is the matrix that double-centers the input matrix by subtracting the row and column means from every element of the matrix. Introducing the constant ![]() creates an equivalence to PCA if
creates an equivalence to PCA if ![]() is a Euclidean distance function:
is a Euclidean distance function:
| (4) |
The eigenvalues ![]() and eigenvectors
and eigenvectors ![]() of
of ![]() are calculated. To reduce to dimension
are calculated. To reduce to dimension ![]() with
with ![]() , only the first
, only the first ![]() of a ranked list of the
of a ranked list of the ![]() and
and ![]() are selected (e.g.
are selected (e.g. ![]() for a two-dimensional mapping).
for a two-dimensional mapping).
The diagonal matrix of the square roots of the eigenvalues ![]() is
is
 |
(5) |
The matrix ![]() of relative coordinates in
of relative coordinates in ![]() dimensions is then calculated by the product of the matrix of eigenvectors
dimensions is then calculated by the product of the matrix of eigenvectors ![]() and the diagonal matrix of the square roots of the eigenvalues
and the diagonal matrix of the square roots of the eigenvalues ![]() :
:
| (6) |
5.2. Case Study: Airport Location Mapping
We begin with one of the original motivating problems related to MDS, reconstructing the relative locations between cities in two dimensions, knowing only the relative distances between each one. We use the United States airport distance data from Section 3 and reconstruct the cities’ relative geographic positions. We begin by squaring the distance functions and calculating the scale of the dataset.
![]()
Next, we create an identity matrix, with all elements being 0 except the diagonal elements, which are 1, and a constant matrix with all elements equal to 1. Both matrices are of the same dimensions and are used in subsequent calculations to transform data from 28 down to two dimensions.
![]()
We then calculate the kernel matrix and double-center the new ![]() -dimensional coordinates.
-dimensional coordinates.
![]()
With the number of dimensions set to 2, the matrix eigenvectors and eigenvalues are calculated. The sign of the first eigenvector is adjusted to lay out the cities from west to east. (Mathematica does not guarantee the direction of an eigenvector.)
![]()

![]()
![]()
The transformation matrix dMTX is calculated from a diagonal matrix formed by the eigenvalues Ev, and the coordinate matrix EmM is calculated from the eigenvectors Em.
![]()

The graph coordinates cMDScoord are calculated and projected to two dimensions.
![]()

Finally, we plot the two-dimensional airport locations in cMDScoord, which shows the airports in the new coordinate system.
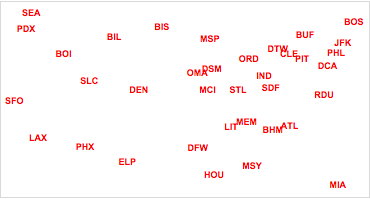
Figure 2. Classical MDS display of relative airport locations.
Classical multidimensional scaling gives a solution that has an excellent projection of the cases in a lower dimension, preserving the between-cases distances from ![]() dimensions. It is important to note, however, that the solution is not unique with respect to the rotation and reflection of the visualization. It is often the case when one knows the “true” orientation of the case locations (e.g. the previous map example), that classical (and metric) MDS methods yield a visualization that is reflected or rotated. In these cases, use of other boundary conditions (fixing known coordinates) helps constrain the orientation of the solution. Often, however, such boundary conditions are not available.
dimensions. It is important to note, however, that the solution is not unique with respect to the rotation and reflection of the visualization. It is often the case when one knows the “true” orientation of the case locations (e.g. the previous map example), that classical (and metric) MDS methods yield a visualization that is reflected or rotated. In these cases, use of other boundary conditions (fixing known coordinates) helps constrain the orientation of the solution. Often, however, such boundary conditions are not available.
Given the above solution, how well does classical MDS reproduce the airport coordinates? Because we have a gold standard, the actual airport locations, comparison of the coordinate estimates is possible. However, the airport locations are represented in a geographic coordinate system, making a comparison of calculated versus actual airport locations a bit more complicated. One approximate way to approach this issue is by first examining the correlation between the actual airport locations, in longitude and latitude coordinates, and the derived coordinates from the classical MDS calculation, using the PearsonCorrelationTest. The units are different, which we will address in a moment, but this does not affect the Pearson correlation test.

Figure 3. Comparison of actual and estimated coordinates.
While the correlations are high, they are not perfect, as we can see by the off-diagonal locations of some of the points. To better visualize these differences, we transform the classical MDS coordinates into longitude and latitude units. This is a transformation that uses the pseudoinverse matrix to translate and rotate to find a good alignment between the two coordinate sets.
![]()
![]()
This type of transformation preserves the relative distances between the classical MDS derived city locations and thus provides a relatively accurate estimate of how close the estimates are to the actual city locations. A more accurate solution would involve mapping of the coordinates to an appropriate spherical projection system; however, this is outside the focus of this article.
The transformation coefficients can now be used to transform the classical MDS derived coordinates into GeoPosition values. To compare the actual (●) with the cMDS calculated (◆) airport locations, we use the GeoListPlot function, which plots both sets of points on a map of North America.
![]()
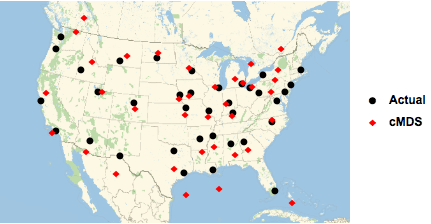
Figure 4. Comparison of airport geo locations.
The accuracy of the classical MDS estimates is fairly good, but not perfect. Part of this error is due to the approximate transformation from arbitrary MDS coordinates to geographic coordinates, and the complex projection to a flat map.
5.3. Function: Classical Multidimensional Scaling
The preceding code can be summarized in a succinct Mathematica function for classical MDS calculation. This function takes a proximity matrix as input and returns coordinates mapped to a space of the specified number of dimensions. The default number of dimensions is two, but the function also works with three or more dimensions.

5.4. Classical Multidimensional Scaling and the Singular Value Decomposition
It is important to note that if the proximity matrix used for classical MDS is formed from a cases by attributes matrix using a Euclidean distance, then classical MDS is equivalent to principal component analysis. Principal component analysis is a specific case of the more general singular value decomposition method (SVD). Details of the SVD mathematics and computational method is not discussed in detail here, but rather we will outline the computational steps necessary to use the method with the function SingularValueDecomposition.
As with classical MDS, a transformation matrix is calculated, and the data is centered to a mean of zero.
![]()
The SVD function is then applied to the normalized data and yields three matrices. Here ww is the diagonal matrix of eigenvalues and vv is the coordinate matrix.
![]()
Next, the two-dimensional projection of the coordinates is calculated.
![]()
When the dataset to be analyzed is very large, then the computational efficiency degrades quickly if the proximity matrix must be directly calculated. In this case, a matrix transformation can be applied to avoid a cell-by-cell proximity matrix calculation; this greatly increases computational efficiency. The computational efficiency for both classical and metric MDS is discussed, with a direct comparison, in Section 7.
5.5. Synopsis: Classical MDS
Classical MDS is a data mining and data exploration method allowing dimensional reduction to be used to highlight possible clusters, similarities, or differences between cases described by high-dimensional attribute vectors. It is a starting point for more rigorous statistical analysis and hypothesis testing. When the input proximity matrix is composed of Euclidean distances, classical MDS is equivalent to both principal coordinate analysis and singular value decomposition. Computational efficiency and possible limitations of classical MDS methods are discussed in Section 7.
6. MDS Methods: Metric Multidimensional Scaling
Metric multidimensional scaling is a second type of MDS. While classical MDS relies on matrix algebra, metric MDS involves computational minimization of the difference between ![]() -dimensional and two-dimensional proximity measures (e.g. Euclidean distance) of the case coordinates in each dimensional system. In essence, the method attempts to minimize the total error between actual and reduced intercase distances for the group of cases as a whole. Metric MDS is also flexible, accommodating different types of proximity metrics (e.g. not Euclidean), as well as different stress functions (nonlinear transformation and distance metrics other than Euclidean) for minimization. This permits nonlinear dimensional reduction, where the projection of data from an
-dimensional and two-dimensional proximity measures (e.g. Euclidean distance) of the case coordinates in each dimensional system. In essence, the method attempts to minimize the total error between actual and reduced intercase distances for the group of cases as a whole. Metric MDS is also flexible, accommodating different types of proximity metrics (e.g. not Euclidean), as well as different stress functions (nonlinear transformation and distance metrics other than Euclidean) for minimization. This permits nonlinear dimensional reduction, where the projection of data from an ![]() -dimensional space to lower dimensions can be done using a nonlinear transformation. The end result is a dimensional reduction that allows for data visualization.
-dimensional space to lower dimensions can be done using a nonlinear transformation. The end result is a dimensional reduction that allows for data visualization.
As in the previous section, we first describe the mathematics with some references to implementation, then demonstrate a practical example.
6.1. Mathematics
Metric MDS also begins with a ![]() proximity matrix
proximity matrix ![]() , which represents the original
, which represents the original ![]() -dimensional proximity measure for the cases
-dimensional proximity measure for the cases ![]() , where
, where ![]() . This proximity matrix can be given de novo, as in distances between airports, or can be calculated from a cases by attributes matrix as described previously,
. This proximity matrix can be given de novo, as in distances between airports, or can be calculated from a cases by attributes matrix as described previously,
 |
(7) |
Initial values for the case coordinates, used to seed the minimization algorithm, can be generated at random or calculated using values from the classical MDS algorithm. So we next set up the initial coordinate matrix ![]() for a two-dimensional projection, for the minimization algorithm,
for a two-dimensional projection, for the minimization algorithm,
 |
(8) |
where ![]() . Next, we create a matrix of variables representing the coordinates that the minimization algorithm estimates:
. Next, we create a matrix of variables representing the coordinates that the minimization algorithm estimates:
 |
(9) |
We then calculate a matrix of distance functions for ![]() using Euclidean distance between any two elements
using Euclidean distance between any two elements ![]() and
and ![]() ,
,
| (10) |
The distance matrix ![]() is now given by
is now given by
 |
(11) |
For the minimization, several different types of stress functions may be used. Here we select the stress function to be the sum of the squares of the errors (SSE):
| (12) |
We use the efficient and robust "DifferentialEvolution" method, with sufficient search points to avoid local minima. Some aspects of the choice of minimization methods are discussed later.
6.2. Case Study: Mapping Antibody Reactivity Differences between Influenza Strains
This example is motivated by the need to determine if an influenza strain is sufficiently different, as determined by the immune system, so that even if a person has prior immunity from vaccination against other similar strains, it will be insufficient to protect against the new strain. Influenza vaccination relies on the immune system’s ability to generate protective antibodies, which are proteins that bind to the virus and block its ability to infect cells in the respiratory system. The major protection comes from antibodies that bind the viral surface protein hemagglutinin, the protein responsible for attaching the virus to the target cells. Variations in the hemagglutinin protein structure between influenza strains allow the viruses to evade the immune system and cause an infection. This is why the influenza vaccine composition is changed every year. Figuring out which strains to include in the influenza vaccine is an annual problem for the international organizations that recommend changes in the seasonal influenza vaccine.
Metric MDS provides a graphical way of visualizing influenza strain similarity, derived from experiments measuring the ability of serum from animals infected with influenza to bind to the target virus hemagglutinin. There has been extensive literature over the last decade on the use of metric MDS for this purpose, referred to as antigenic cartography [8–11]. Metric MDS was chosen for dimensional reduction and visualization by several groups, as classical MDS methods could not be easily adapted to solve several issues [8, 9]. These included the need for complete datasets [10, 11], where in some cases data was missing due to experimental considerations. Metric MDS methods could be adapted to impute relationships [10]. In addition, metric MDS was viewed as a more accurate estimator of influenza strain antigenic distance due to correlations with the binary logarithm of the hemagglutinin inhibition assay serum titers [8, 9]. Use of metric MDS continues in the influenza literature [10, 11], although newer methods of measuring antibody reactivity do not have the same issues as older assays, and classical MDS could be used to the same end. To illustrate the method of metric MDS, however, we will use it in this example. We discuss method selection in Section 7, as well as considerations of computational efficiency.
We begin with the proximity matrix derived in the previous section from the antibody reactivity to influenza hemagglutinin data. We next calculate the relative positions of each influenza strain with respect to the entire set by minimizing a stress function. The Array function is used to create an array of variables that specify the coordinates for each case as a 2D set of points.
![]()
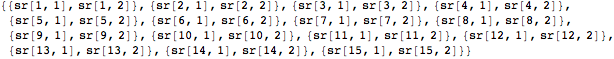
Each sr variable pair will hold a pair of coordinates in two-dimensional space. Finally, we flatten the list of variable pairs to input into the function NMinimize.
![]()
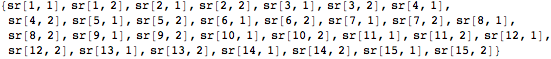
The next section of code creates a series of Euclidean distance calculations for the distances between all combinations of case locations, using the estimated coordinates for each point. For succinctness, only one function within the full matrix is displayed.
![]()
![]()
The stress function is a very large least-squares expression.
![]()
The first nonzero element of the stress function is shown here. The reader may expand the formula to see the entire function, if desired.
![]()
![]()
Finally, we must choose the method for minimizing the stress function. We discuss two options, FindMinimum and NMinimize. A known issue with metric MDS is the existence of local minima, and thus identifying a global minimum cannot be guaranteed [7]. While FindMinimum may be computationally more efficient, it also lacks the ability to specify the number of search points and is prone to finding local minima instead of global minima. These issues are discussed in some detail in Section 7. In contrast, the NMinimize function allows specification of "SearchPoints" to address this issue and may be substantially more robust. For these reasons, we chose NMinimize to optimize the stress function.
![]()

The viral strains are then plotted in the two-dimensional space and are color-coded for clarity (H1, H3, and H5). Note that the coordinate system is arbitrary, in the sense that what is preserved and important are the relative distances between the data points. Thus, we have omitted the axes, which have the same scale in each dimension.
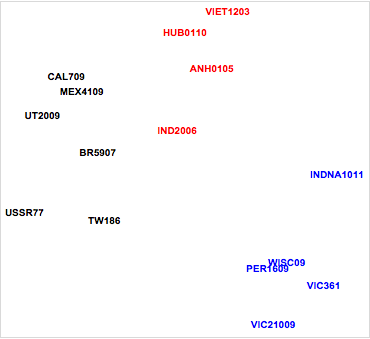
Figure 5. Relative antigenic distances for influenza strains.
Examining the plot, it is immediately apparent that the influenza strains fall into four distinct clusters. Two of these correspond to major hemagglutinin protein strains (H3, H5). In addition, we can see differences between temporally distinct strains in the H1 influenza strains, giving perhaps two clusters. For example, note the antigenic distance between the pandemic A/California/07/2009 (CAL709), A/Utah/20/2009 (UT2009), and A/Mexico/41/2009 (MEX4109) strains and the other H1 influenza strains. This likely reflects molecular mutations in the hemagglutinin proteins for the 2009 strains. These mutations resulted in decreased binding of antibodies from ferrets infected with earlier influenza strains. This finding was consistent with the decreased population immunity observed in humans to A/California/07/2009 and demonstrates the pandemic nature of that particular influenza strain.
Given that the influenza strains seem to cluster together, what is the relationship between MDS and methods of unsupervised data clustering used in data mining? The answer lies in the use of the proximity matrix, which is used by both hierarchical and agglomerative clustering methods to determine relatedness. This relationship was noted by the developers of the MDS methods [1, 2, 8]. To briefly demonstrate, we apply hierarchical clustering to the same proximity matrix Dm, using the DirectAgglomerate function. Ward’s minimum variance method is used for determining cluster linkage.
![]()
The resulting dendrogram is displayed in Figure 6. Note the grouping of the different virus types (H1, H3, and H5) based on the reactivity of ferret serum after infection with a single virus and the resulting antibodies against the hemagglutinin proteins.

Figure 6. Dendrogram of influenza virus relationships.
To explore which viruses are antigenically closer to each other, a heat map of the relative distances from the calculated coordinates (or the original proximity matrix) can then be created with DistanceMatrix and ArrayPlot. Note that more antigenically similar influenza strains have smaller distances between each other.
![]()
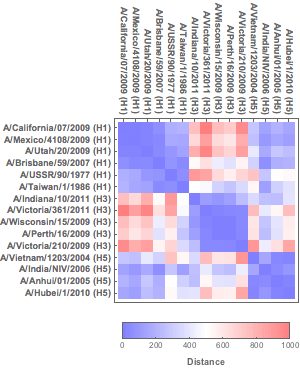
Figure 7. Heat map of proximity matrix data.
6.3. Function: Metric Multidimensional Scaling
As in the previous section, we end with a general function for the metric MDS calculation. The function requires several input variables, including precision, accuracy, the minimization method, and the maximum number of iterations. This function uses the classical MDS routine for the initial coordinate estimates. Some suggested defaults are given in the function definition to be passed to NMinimize. The metric MDS function is written for two-dimensional mapping that could easily be generalized for three dimensions.

6.4. Nonlinear Stress Functions
One of the advantages of metric MDS methods is the ability to use nonlinear mappings from ![]() -dimensional to visualizable space. We now use this function to create an MDS map comparing the influenza hemagglutinin proteins by sequence similarity. Recall that the proximity measure for sequence comparison is the Smith–Waterman sequence distance, which is not a Euclidean distance function. Thus, this violates the assumptions of classical MDS and makes metric MDS the appropriate method to use, albeit at computational cost. Taking the hemagglutinin protein proximity matrix, fluPm, we apply the metricMDS function and obtain the coordinates. We also apply hierarchical clustering to the proximity matrix fluPm.
-dimensional to visualizable space. We now use this function to create an MDS map comparing the influenza hemagglutinin proteins by sequence similarity. Recall that the proximity measure for sequence comparison is the Smith–Waterman sequence distance, which is not a Euclidean distance function. Thus, this violates the assumptions of classical MDS and makes metric MDS the appropriate method to use, albeit at computational cost. Taking the hemagglutinin protein proximity matrix, fluPm, we apply the metricMDS function and obtain the coordinates. We also apply hierarchical clustering to the proximity matrix fluPm.
One of the advantages of metric MDS methods is that one can use nonlinear stress functions to emphasize particular regions of data relationships. One of the first nonlinear metric MDS methods was that of Sammon’s mapping [12, 13]. Sammon’s mapping can be useful in revealing underlying data structure or differences with nonlinear relationships. This mapping minimizes the following general nonlinear form of stress functions:
| (13) |
where ![]() is a weighting function and
is a weighting function and ![]() is a constant. In the case of Sammon’s mapping, the stress function is defined by
is a constant. In the case of Sammon’s mapping, the stress function is defined by ![]() ,
, ![]() , with
, with ![]() specified as [12]:
specified as [12]:
| (14) |
Many related nonlinear mappings exist of the same form [12, 14]. For the purposes of this example, we explore applying a nonlinear exponential MDS mapping with a stress function defined by ![]() ,
, ![]() ,
, ![]() ,
, ![]() , and
, and ![]() :
:
| (15) |
Note that ![]() is an empirically specified tuning factor. This nonlinear mapping function decreases the contribution to the overall stress function of larger
is an empirically specified tuning factor. This nonlinear mapping function decreases the contribution to the overall stress function of larger ![]() and has the effect of expanding the mapped distances between data points with smaller
and has the effect of expanding the mapped distances between data points with smaller ![]() . The advantage of this mapping is that the weight of any point in the minimization is inversely proportional to its magnitude. Thus, smaller differences between data elements are spread out. The coding of the exponential MDS function lets you specify
. The advantage of this mapping is that the weight of any point in the minimization is inversely proportional to its magnitude. Thus, smaller differences between data elements are spread out. The coding of the exponential MDS function lets you specify ![]() .
.

To demonstrate, we apply the metric nonlinear MDS method to the above fluPm dataset of protein sequence comparisons. First, we apply the standard metric MDS function defined in the previous section, metricMDS, and generate a dendrogram to highlight the sequence differences.
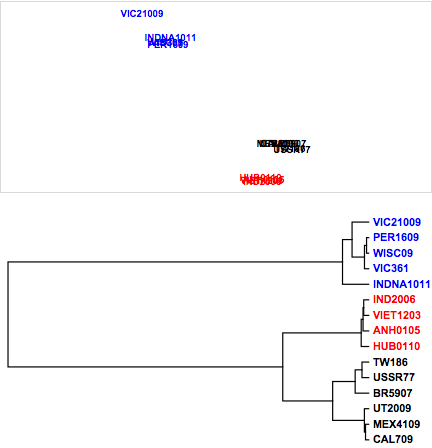
Figure 8. Influenza virus relationships from protein sequences.
While the major influenza viral subtypes (H1, H3, and H5) all cluster together, minimal differences can be observed with respect to sequences differences within each subtype. Using the nonlinear mapping, as shown in Figure 9, accentuates the small differences between the hemagglutinin protein sequences. Note the negative exponential weighting function, with the large distances between hemagglutinins being given less weight in the minimization. We are now able to visualize the division of the H1 influenza hemagglutinins into two major clusters and the split between clusters of the H3 substrains within the visualization. It is worth noting that the hierarchical clustering may capture these differences.
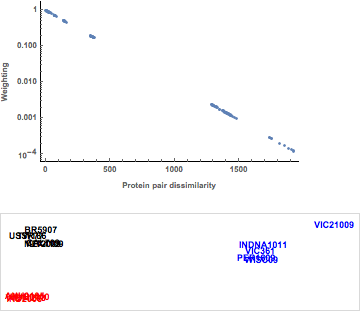
Figure 9. Weighting function and nonlinear mapping of protein sequences.
6.5. Synopsis: Metric MDS
Metric multidimensional scaling is a more flexible method compared to classical MDS. While computationally less efficient, it allows nonlinear dimensional reduction that is not possible with the SVD or PCA method of classical MDS. This functionality can be used to highlight possible clusters, similarities, or differences between cases described by high-dimensional attribute vectors and to weight or penalize the stress function based on data attributes. As with classical MDS, complementary visualization methods allow for a fuller picture of case differences but must be carefully interpreted when nonlinear stress functions are used.
7. Some Considerations on Selecting a Method for MDS
From a computational perspective, several features of classical and metric MDS methods should be considered when selecting which method to use for a specific analysis.
Classical MDS is computationally straightforward but has some subtle mathematical constraints. For a solution to exist in Euclidean space, the distance matrix must follow a set of necessary and sufficient conditions [1, 15]. In practice, this is rarely an issue. When it is an issue, it often manifests by a computational result expressed in imaginary numbers. If, however, the proximity matrix is composed of Euclidean distances, classical MDS becomes principal component analysis, and the support vector machine (SVM) function can be used. This is much more computationally efficient, as we demonstrate in Section 7.3, and is the preferred method of MDS for visualization of most data.
Metric MDS has somewhat more relaxed constraints than classical MDS. For very high numbers of initial dimensions ![]() , metric MDS is computationally and memory intensive. This is primarily due to the need to use iterative optimization to minimize the stress function, whereas classical MDS uses more efficient matrix algebra operations. In addition, one needs to pay attention to the algorithms used to perform metric MDS to ensure that minimization finds global rather than local minima.
, metric MDS is computationally and memory intensive. This is primarily due to the need to use iterative optimization to minimize the stress function, whereas classical MDS uses more efficient matrix algebra operations. In addition, one needs to pay attention to the algorithms used to perform metric MDS to ensure that minimization finds global rather than local minima.
We next examine the selection of minimization algorithms for metric MDS, compare the computational efficiency of classical and metric MDS methods, and discuss the particular circumstances where metric MDS may be the method of choice.
7.1 Convergence, Accuracy, and Selection of Minimization Algorithms for Metric MDS
With metric MDS, selecting a minimization algorithm with an appropriate Method setting for Mathematica’s built-in function NMinimize may require some investigation. In addition, a known issue with metric MDS is the existence of local minima, so that a global minimum cannot be guaranteed [7]. Using a large number of starting points or selecting the appropriate minimization function and method may help. In this article we have used NMinimize with Method set to the "RandomSearch" option. We also specified sufficient search points to avoid local minima. This can be an important issue when no other external data is available to constrain the solution and when it is not known if other minimization methods will routinely converge to a solution.
It is worth exploring this tradeoff between convergence, accuracy, and computational efficiency. For example, the execution time of NMinimize increases with the number of initial search points. The next example uses NMinimize on the influenza antibody dataset distance matrix Dm and the method "RandomSearch". In this case, however, the number of search points appears to make little difference in the residual after optimizing the stress function.



Figure 10. Efficiency of NMinimize (RandomSearch).
In the case of RandomSearch, there appears to be no tradeoff with this particular dataset. In contrast, the theoretically more robust setting ![]() finds a small number of varying minima, which improves after specifying a larger number of search points. Also note the improved execution times, which are an order of magnitude less than with RandomSearch.
finds a small number of varying minima, which improves after specifying a larger number of search points. Also note the improved execution times, which are an order of magnitude less than with RandomSearch.

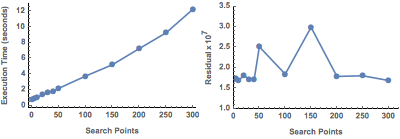
Figure 11. Efficiency of NMinimize (DifferentialEvolution).
Another consideration is the speed of computation. The FindMinimum function is often much faster than NMinimize, but the tradeoff is the risk that the algorithm will not converge or that it will find local instead of global minima. For the influenza data, FindMinimum can suffer from both nonconvergence and local minima, although the former issue may be sporadic. The sporadic nature of convergence can be frustrating and problematic. The issue of finding local minima is potentially more serious. To explore this further, we minimized 300 times with FindMinimum.

The results are displayed as a histogram in Figure 13. We found that a number did not converge with the influenza antibody-reactivity dataset. Of even more concern is that FindMinimum was not consistent, and often appeared to provide local rather than global minima. This may in some cases be dataset specific. The starting points selected, here a RandomReal number between ![]() and
and ![]() for each coordinate, are also critical for FindMinimum.
for each coordinate, are also critical for FindMinimum.

Figure 12. Residuals and convergence using FindMinimum.
The convergence can be improved by minimizing the sum of the squares rather than the Euclidean distance. Note that the minimum residual is higher than when minimizing the sum of the differences between distances rather than the square of the difference between distances. This may be due to computational considerations, in that there is differentiability at the optima when minimizing the sum of the squares of the distances, while this is lacking if minimizing the difference between the distances. Still, a moderate number (2% to 15% from 300 attempts) of the solution attempts did not converge or find global minima. Whether this is an issue for your particular application depends on both the level of precision required and the computational efficiency required.
![]()
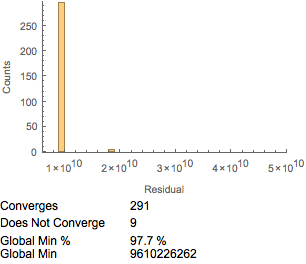
Figure 13. FindMinimum and minimizing the sum of the squares.
Given the low percentage of nonconvergence and the presence of local minima, one could execute FindMinimum several times and take the results that converge with the lowest minimum. This does increase the time for execution. An alternative is to use the function NMinimize. The tradeoff is an increased computational time for consistent convergence on global minima, as shown here. This demonstration takes several minutes to perform on this dataset, even with parallelization.


Figure 14. Residuals and convergence using NMinimize.
A single minimum residual is obtained for all 300 separate runs of NMinimize. Thus, unless local minima are known not to be an issue, NMinimize may be a more consistent minimization method for metric MDS routines.
7.2. Computational Efficiency
The issues of optimization and computational efficiency make it useful to benchmark classical and metric MDS methods with respect to execution time. Therefore, we compared SVD with classical and metric MDS across the three example datasets. For metric MDS, we include both FindMinimum and NMinimize. The measured execution times encompass all the steps from the input of the proximity matrix through final coordinate output, but not plotting. The previously created classicalMDS and metricMDS functions defined previously were used, along with new functions created for singular value decomposition (mdsSVD) and metric MDS using FindMinimum (findMinMDS).


![]()
![]()
Note that FindMinimum failed to converge for the airport dataset but did converge for both the flu antibody and sequence datasets, a behavior that may vary between executions. Timings are shown here.

Overall, there is a speed advantage to using classical MDS and SVD, which are several orders of magnitude faster than metric MDS using either FindMinimum or NMinimize. This advantage assumes that the proximity matrix can be formulated using a Euclidean distance, and thus other measurements (e.g. Manhattan distance, etc.) may require other methods. It also assumes that you require a linear mapping from the ![]() -dimensional space to the visualizable space. SVD has the advantage that, for many applications, the proximity matrix may not have to be directly calculated, saving additional memory and computational time [8]. With respect to metric MDS, there are also tradeoffs. For moderately large datasets, if one can optimize the conditions to avoid nonconvergence and local minima, FindMinimum is a good choice. Note that the execution time for FindMinimum in the airport and influenza antibody dataset examples occurred in the setting of failure to converge. Although slower, metric MDS using NMinimize had no issues with nonconvergence or finding local instead of global minima.
-dimensional space to the visualizable space. SVD has the advantage that, for many applications, the proximity matrix may not have to be directly calculated, saving additional memory and computational time [8]. With respect to metric MDS, there are also tradeoffs. For moderately large datasets, if one can optimize the conditions to avoid nonconvergence and local minima, FindMinimum is a good choice. Note that the execution time for FindMinimum in the airport and influenza antibody dataset examples occurred in the setting of failure to converge. Although slower, metric MDS using NMinimize had no issues with nonconvergence or finding local instead of global minima.
7.3. Some Thoughts on Method Selection: Classical versus Metric MDS
Given the computational advantages of classical MDS in its various forms (principal component analysis, singular value decomposition, etc.), why would you choose metric MDS, which requires numerical optimization? For those wishing an in-depth and lucid explanation, we refer you to the initial work of Gower [15] and the outstanding article by Shiens [16]. In short, classical MDS methods are usually preferable to and always computationally more efficient than metric MDS methods. The method is computationally efficient and robust; there are very few cases where classical MDS methods fail to provide a visualization. For some datasets, classical MDS methods may not provide optimal visualization, and metric MDS should be considered. Some examples include datasets whose variables have a non-Gaussian distribution, those where the distance metric is not Euclidean, those requiring imputation of missing data [10, 11], or cases with a parametrization in a nonorthogonal coordinate system [16]. For nonlinear dimensional reduction, which is often used in graph layout algorithms, metric MDS with stress function minimization is the method of choice [14].
Thus, selection of an MDS method for visualization should consider several factors. If the distance function is Euclidean, and especially if the dataset is large, SVD or classical MDS are the most appropriate and computationally efficient methods. In the less common case where metric MDS is used, careful consideration should be given to the choice of stress function minimization method (in this example NMinimize versus FindMinimum) to avoid local minima.
8. Summary and Conclusion
We have demonstrated the application of two methods of multidimensional scaling, classical and metric, for the visualization of similarity/proximity of high-dimensional data, with reduction to two or three dimensions. We have shown how these methods can be used to visualize the relatedness of influenza virus strains with respect to antibody-mediated immunity, as well as their utility in reconstructing relative spatial-geographic locations using only interlocation distances. These MDS methods are, however, quite generalizable. Both classical and metric MDS rely on a proximity matrix. While the examples in this article use continuous variables and sequences or case attributes, a variety of data types with appropriate proximity metrics can be visualized with MDS methods, such as molecular DNA or protein sequence data (Smith–Waterman similarity, Needleman–Wunsch similarity, or Damerau–Levenshtein distance), Boolean data (Hamming distance), and images (image distance, color distance). For cases with a single data attribute (e.g. sequence similarity, distance), no data scaling is necessary. For cases with multiple attributes having disparate units, standardization (e.g. ![]() -score) and rescaling are needed to equally weight each attribute.
-score) and rescaling are needed to equally weight each attribute.
In our examples, we used reduction down to two dimensions for multidimensional data visualization. Reduction to three dimensions is easily accomplished with both classical and metric MDS, although the computational cost may increase with the added dimension, depending on the method used. While we have emphasized data visualization, the classical MDS method can also be used for reduction to four or more dimensions as a method of identifying the weighted combinations of components that contribute the most to the data variance. In these cases, the goal is to select a small set of variables that explain the most variance in the dataset, such that this minimum set of variables can be further used for statistical or other modeling.
We have also shown how MDS methods are related to clustering algorithms, which also use proximity matrices to compare and classify cases by their attributes. This relationship also allows creative graphical display of multidimensional data from several vantage points. For example, one can use the MDS plot to display the relative proximity of cases to each other and plot marker coloring or other methods to add information regarding other case attributes. Some caution is in order, however, as different proximity measures and data transformations may give different clustering and classification. Parallel display of dendrograms and heat maps may also enhance understanding of the relationship of data clusters to each other. Similarly, heat maps, combined with MDS displays, are particularly helpful for data exploration, in that they enhance visual identification of those data attributes (dimensions) that contribute the most to differentiating between case clusters.
Care should be taken when selecting the MDS method. In most cases, classical MDS will be the most computationally efficient method, especially for very large datasets. In the cases where metric MDS is optimal, such as the use of nonlinear mapping, care should be taken to choose a minimization method that is robust and avoids local minima. Performing some testing on a subset of the data can be very informative regarding convergence, accuracy, and computational efficiency. While we did not discuss in detail how constraining optimization problems can improve computational efficiency and accuracy, this should also be considered whenever boundary conditions or other information is available.
Finally, one must remain aware that these methods reveal only associative patterns. Further analysis with rigorous statistical inference methods is needed to test the validity and specify the error boundaries of these associations. Mechanistic studies should be performed, if possible, to confirm suspected causal relationships. Overall, however, MDS methods are excellent for dimensional reduction and data exploration with the goal of creating comprehensible and informative quantitative graphical representations of multidimensional data.
Acknowledgments
We would like to thank Ollivier Hyrien, Alex Rosenberg, Chris Fucile, and Melissa Trayhan for their suggestions and critical reading of the manuscript. We would also like to acknowledge the reviewer at the Mathematica Journal, who suggested several improvements and additions, including the evaluation of execution time and accuracy for the MDS methods, and the discussion regarding the relative merits of SingularValueDecomposition, NMinimize, and FindMinimum. This work was supported by grants to the authors from the Philip Templeton Foundation, as well as the National Institute of Allergy and Infectious Diseases and the National Institute of Immunology, grant and contract numbers HHSN272201000055C, AI087135, AI098112, AI069351.
References
| [1] | J. C. Gower, “Some Distance Properties of Latent Root and Vector Methods Used in Multivariate Analysis,” Biometrika 53(3–4), 1966 pp. 325–338. doi:10.1093/biomet/53.3-4.325. |
| [2] | W. S. Torgerson, “Multidimensional Scaling: I. Theory and Method,” Psychometrika, 17(4), 1952 pp. 401–419. doi:10.1007/BF02288916. |
| [3] | W. S. Torgerson, Theory and Methods of Scaling, New York: Wiley, 1958. |
| [4] | T. F. Cox and M. A. A. Cox, Multidimensional Scaling, Boca Raton: Chapman and Hall, 2001. |
| [5] | I. Borg, P. Groenen, Modern Multidimensional Scaling: Theory and Applications, 2nd ed., New York: Springer, 2005 pp. 207–212. |
| [6] | M. Wish and J. D. Carroll, “Multidimensional Scaling and Its Applications,” Handbook of Statistics, Vol. 2: Classification, Pattern Recognition and Reduction of Dimensionality (P. R. Krishnaiah and L. N. Kanal, eds.), Amsterdam: North-Holland, 1982 pp. 317–345. |
| [7] | V. Saltenis, “Constrained Optimization of the Stress Function for Multidimensional Scaling,” in Computational Science–ICCS 2006 (V. Alexandrov, G. van Albada, P. A. Sloot, and J. Dongarra, eds.), Berlin: Springer, 2006 pp. 704–711. link.springer.com/chapter/10.1007/11758501_ 94. |
| [8] | D. J. Smith, A. S. Lapedes, J. C. de Jong, T. M. Bestebroer, G. F. Rimmelzwaan, A. D. M. E. Osterhaus, and R. A. M. Fouchier, “Mapping the Antigenic and Genetic Evolution of Influenza Virus,” Science, 305(5682), 2004 pp. 371–376. doi:10.1126/science.1097211. |
| [9] | T. Bedford, M. A. Suchard, P. Lemey, G. Dudas, V. Gregory, A. J. Hay, J. W. McCauley, C. A. Russell, D. J. Smith, and A. Rambaut, “Integrating Influenza Antigenic Dynamics with Molecular Evolution,” eLife, 3:e01914, 2014. doi:10.7554/eLife.01914. |
| [10] | Z. Cai, T. Zhang, and X.-F. Wan, “Concepts and Applications for Influenza Antigenic Cartography,” Influenza and Other Respiratory Viruses, 5(Suppl. s1), 2011 pp. 204–207. www.ncbi.nlm.nih.gov/pmc/articles/PMC3208348. |
| [11] | D. W. Fanning, J. A. Smith, and G. D. Rose, “Molecular Cartography of Globular Proteins with Application to Antigenic Sites,” Biopolymers, 25(5), 1986 pp. 863–883. doi:10.1002/bip.360250509. |
| [12] | S. Lespinats and M. Aupetit, “False Neighbourhoods and Tears are the Main Mapping Defaults. How to Avoid It? How to Exhibit Remaining Ones?,” QIMIE/PAKDD 2009 (PAKDD Workshops, Thailand), Bangkok, 2009 pp. 55–64. conferences.telecom-bretagne.eu/data/qimie09/lespinats_aupetit_QIMIE _ 2009.pdf. |
| [13] | J. W. Sammon, “A Nonlinear Mapping for Data Structure Analysis,” IEEE Transactions on Computers, C-18(5), 1969 pp. 401–409. doi:10.1109/T-C .1969.222678. |
| [14] | L. Chen and A. Buja, “Stress Functions for Nonlinear Dimension Reduction, Proximity Analysis, and Graph Drawing,” Journal of Machine Learning Research, 14(1), 2013 pp. 1145–1173. dl.acm.org/citation.cfm?id=2502616. |
| [15] | J. C. Gower, “A Comparison of Some Methods of Cluster Analysis,” Biometrics, 23(4), 1967 pp. 623–637. doi:10.2307/2528417. |
| [16] | J. Shiens, “A Tutorial on Principal Component Analysis.” arxiv.org/pdf/1404.1100.pdf. |
| M. S. Zand, J. Wang, and S. Hilchey, “Graphical Representation of Proximity Measures for Multidimensional Data,” The Mathematica Journal, 2015. dx.doi.org/doi:10.3888/tmj.17-7. | |
About the Authors
Martin S. Zand is a professor of medicine and director of the Rochester Center for Health Informatics at the University of Rochester. His research includes the application of informatics, graph theory, and computational modeling to vaccine immunology, gene regulatory networks, and health care delivery.
Jiong Wang is a molecular virologist and research assistant professor at the University of Rochester. She works on influenza vaccination responses and developing high-dimensional measurements of vaccine immune responses.
Shannon Hilchey is a cellular immunologist and research associate professor at the University of Rochester. He studies human immune responses to vaccination, hematologic cancers, and organ transplants in human subjects.
Martin S. Zand, MD, PhD
University of Rochester Medical Center
601 Elmwood Avenue – Box 675
Rochester, NY 14618
martin_zand@urmc.rochester.edu
Jiong Wang, PhD
University of Rochester Medical Center
601 Elmwood Avenue – Box 675
Rochester, NY 14618
jiong_wang@urmc.rochester.edu
Shannon Hilchey, PhD
University of Rochester Medical Center
601 Elmwood Avenue – Box 675
Rochester, NY 14618
shannon_hilchey@urmc.rochester.edu